Você não leu o título errado. Este é o primeiro post de uma série tendo Bioinformática como temática central. Daqui para frente este será um tema recorrente no blog do Assert Labs como uma forma esboçar uma série de estudos introdutórios que nos tem ajudado a imergir neste novo mundo.
O objetivo deste post em específico é apresentar ferramentas para a busca de moléculas de RNA na Internet, a predição da estrutura secundária da molécula de RNA e encontrar RNAs não codificantes.
Estruturas secundárias
Uma molécula de RNA de estrutura simples não estará estável enquanto algumas das suas bases nitrogenadas não estiverem protegidas de contato com seu solvente natural - a água. A maneira mais efetiva de fazer isso é unindo as bases nitrogenadas em pares. Esta ligação entre pares de bases é mais efetiva quando uma guanina (G) é ligada a uma citosina (C) em comparação com uma ligação entre uma uracila (U) ligada com uma adenina (A).
O emparelhamento dessas bases forma a estrutura secundária do RNA. Quando uma molécula de RNA possui dois longos trechos complementares, ela “dobra“ para formar os pares de bases resultando em uma haste de ligações estáveis. Estas hastes podem ser imperfeitas e conter regiões arredondadas de bases não pareadas (ver figura abaixo).
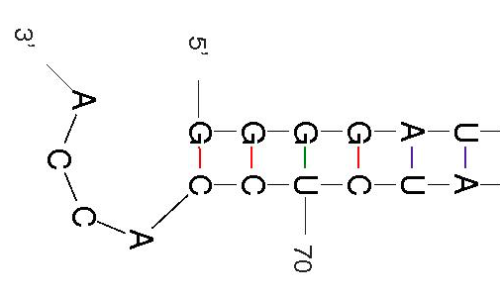
Existe uma tendência natural para a molécula de RNA buscar ter a estrutura secundária mais estável a partir do melhor conjunto de ligações de bases possível de ser formado. O conceito lowest-energy model (modelo de menor quantidade de energia) preconiza que as estruturas de RNA estáveis sempre possuem um valor energético negativo em Kcal/mol. Portanto, para “desdobrar” uma sequencia de RNA desfazendo as ligações entre as bases nitrogenadas é necessário prover energia (calor) a essas moléculas. A estabilidade de um RNA dobrado depende não só da quantidade de ligações GC (GC content), mas da quantidade de ligações de uma maneira geral e do tamanho das regiões arredondadas, também chamadas de regiões de loop. Algoritmos sofisticados como o mfold podem levar essas especificidades em conta para predizer qual seria uma dobra de estabilidade ótima dada a estrutura primária do RNA.
Infelizmente essas predições da forma secundária do RNA não são totalmente confiáveis. Essa predição exige que se pressuponha que o RNA dobre-se em si mesmo dentro de uma célula, o que nem sempre é verdade uma vez que existe interações terciárias e efeitos de determinadas proteínas, íons, outros RNAs e outros elementos químicos sobre a dobra. Essas interações terciárias que afetam a estabilidade da dobra do RNA incluem pseudo-knotes que são longas sequencias de ligações entre um loop e uma outra porção da molécula de RNA.
Por conta disto, nem sempre é verdade que a predição de uma estrutura secundária esteja totalmente correta. A regra geral é que as características mais energeticamente estáveis tendam a ser razoavelmente próximas da verdade quando passam pelo processo de predição.
Tipos de RNA
Os RNAs podem ser divididos em dois grupos: 1. RNAs que podem ser transformados em proteínas devido ao trabalho dos ribossomos e 2. RNAs que são utilizados diretamente pelas células.
RNAs não codificantes auxiliam as células nos processos de transcrição, seccionamento e e replicação e outras atividades. Com exceção do RNA ribossomal esses RNAs tendem a ser curtos e pobremente conservados. Um integrante deste grupo é o tRNA (transport RNA) responsável pela montagem de proteínas a partir do transporte de aminoácidos. Existe ao menos um tipo de tRNA para cada aminoácido.
O fenômeno conhecido como RNA inference (RNAi) em células eucariontes ocorre quando pequenas porções de RNA não codificantes interferem no RNA codificante prevenindo a expressão de ao menos um gene. Um tipo de RNA que provoca esse fenômeno é o silencing RNA (siRNA), descoberto inicialmente em plantas que tem a habilidade de inativar (silenciar) um gene.
A descoberta do siRNA abriu as portas para a observação de outras pequenas porções de RNA que são chamadas de micro-RNA (miRNA). Pouco se sabe a respeito do miRNA e sua função nas células e a busca e identificação destes é bastante difícil devido ao seu tamanho reduzido em comparação com o genoma. Sabe-se apenas que o siRNA diferencia-se por ter uma cadeia dupla e o miRNA possui cadeia simples.
Mfold
Mfold é uma implementação do algoritmo de Zuker que tem a função de prever a estrutura secundária energeticamente ótima de um RNA. Para tanto, o algoritmo utiliza um modelo que leva em conta características físicas, como pH e temperatura, que afetam a dobra do RNA. Este modelo energético ignora interações 3-D ou interações entre proteínas e RNA que podem tornar estáveis uma dobra considerada como não ótima.
Para ajudar no processo de decisão o mfold oferece como resultado modelos não ótimos e deixa na mão do especialista o julgamento da estabilidade da dobra ótima. É possível ainda, a partir dessa implementação, forçar uma interação que é dada como correta.
O mfold pode ser encontrado neste endereço tanto para download quanto para uso on-line. Em ambos os casos ele recebe como parâmetros para a predição da estrutura secundária do RNA a sequência de bases nitrogenadas no formato fasta e os parâmetros do modelo energético.
tRNAScan e PatScan
tRNAs são difíceis de serem encontrados em um genoma. Todos os genomas bacteriais e de eucariontes contém tRNA. Mas por serem pequenos necessitam de ferramentas de pesquisa em bases de dados mais sofisticadas que o BLAST.
Uma ferramenta que pode ser utilizada para o propósito de busca de tRNAs em genomas é o tRNAScan. tRNAScan é ótimo para pesquisas interessadas em tRNAs de uma maneira geral. No entanto, se o interessa da pesquisa está em uma família específica de tRNA que permita a busca por estrutura secundária a ferramenta PatScan é mais adequada. Outra alternativa é utilizar o RNAmot que tem as mesmas funcionalidade que o PatScan.